How to prepare inputs
Geometry
Crystal Structures
| Material | Lattice | Stacking | Exp. Param. | Brillouin Zone | Band Structure | Gap | Eg [eV] |
|---|---|---|---|---|---|---|---|
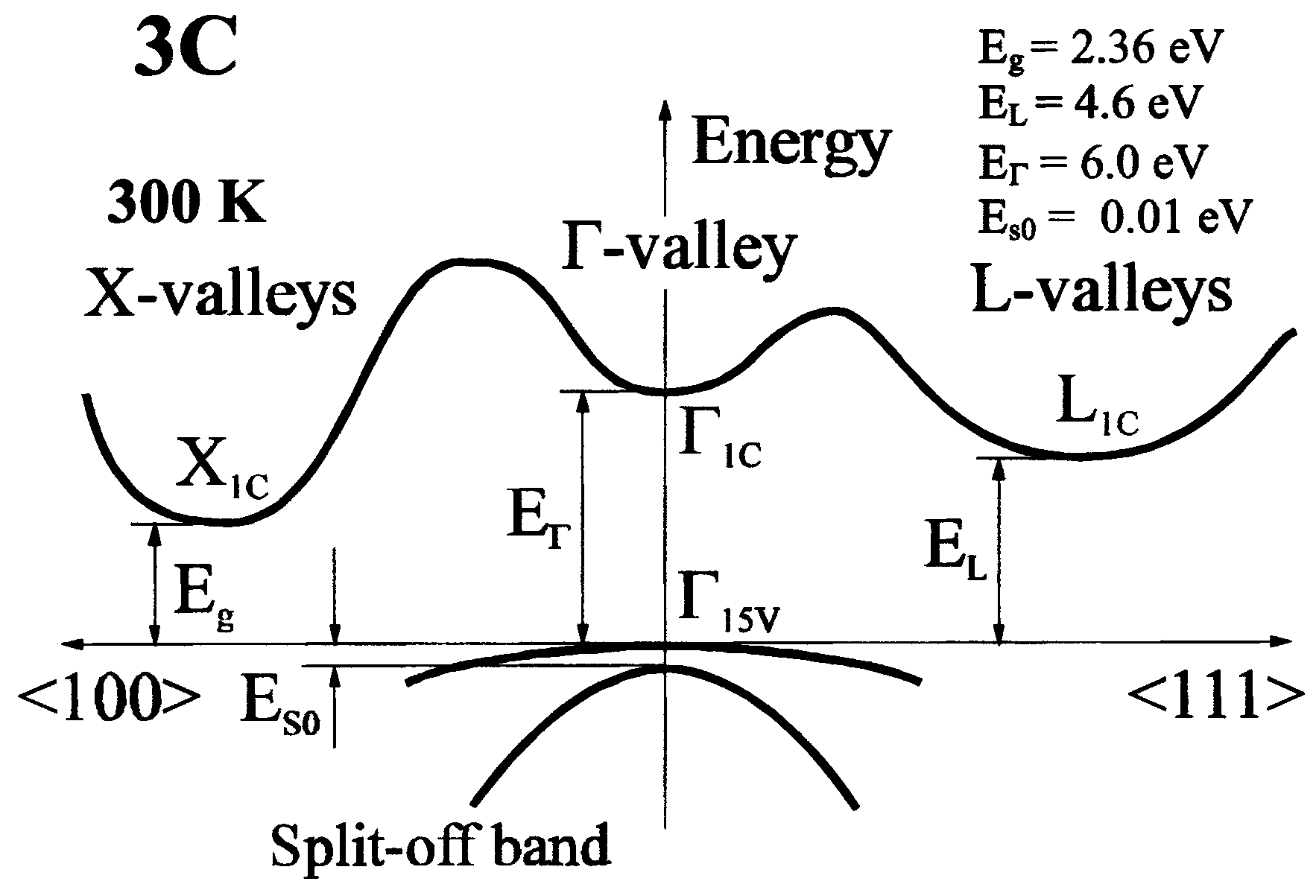
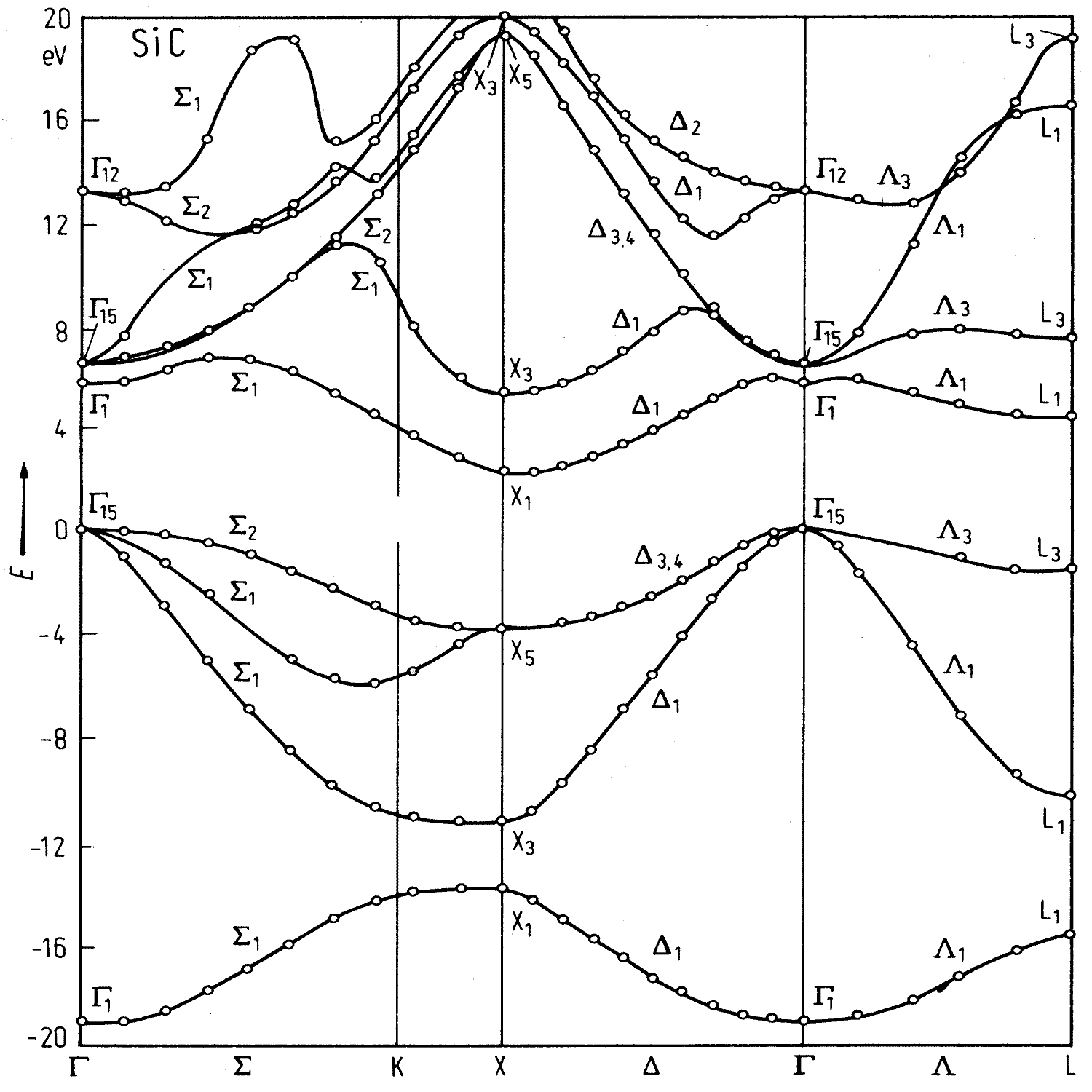
| 3C SiC | Zincblende | ABC | ioffe.ru | FCC (cubic) | PL97 HF74 | Γ - X | 2.36 (300K) |
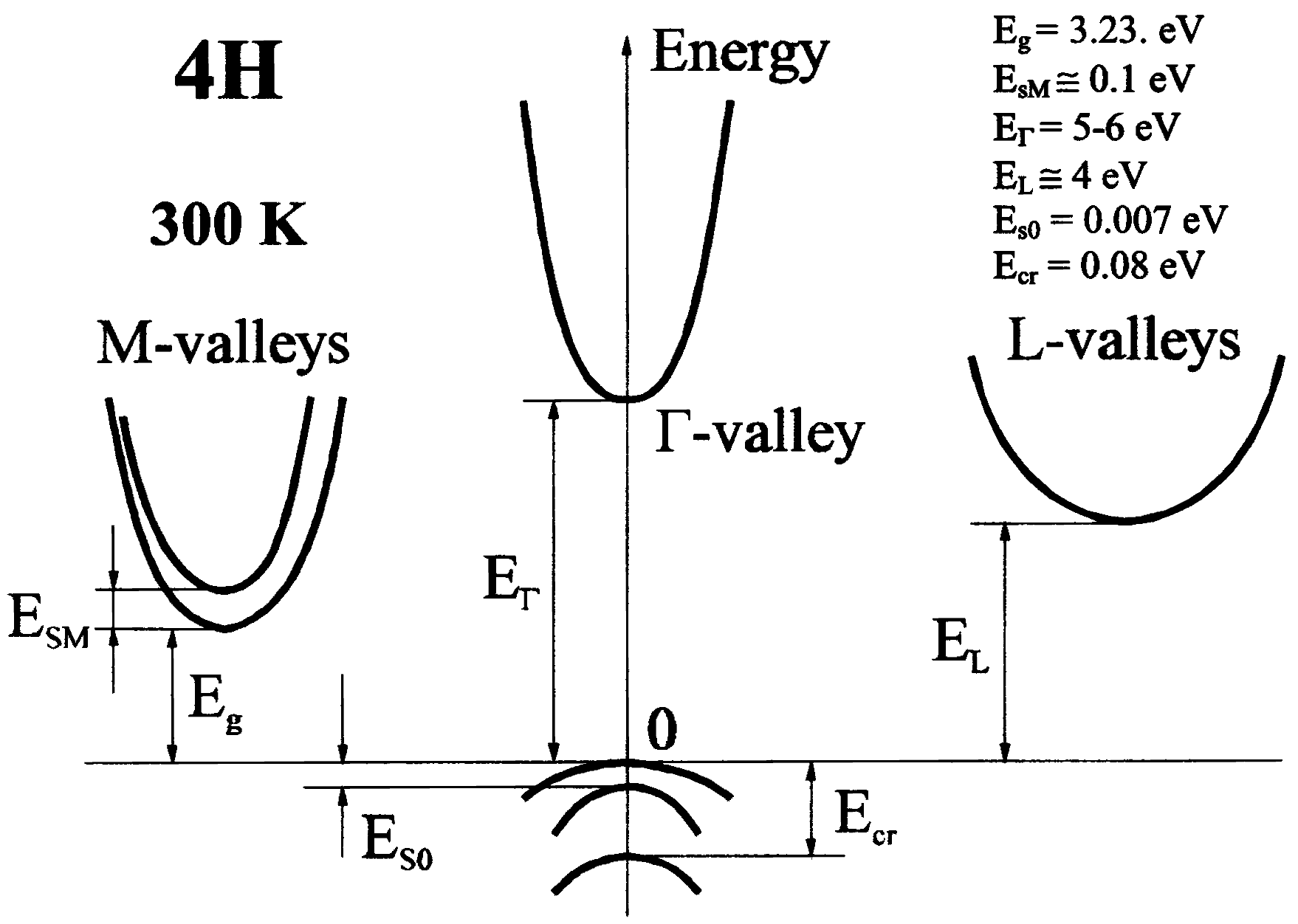
| 4H SiC | Moissanite-4H | ABAC | ioffe.ru | Hexagonal | PL97 | Γ - M | 3.23 (300K) |
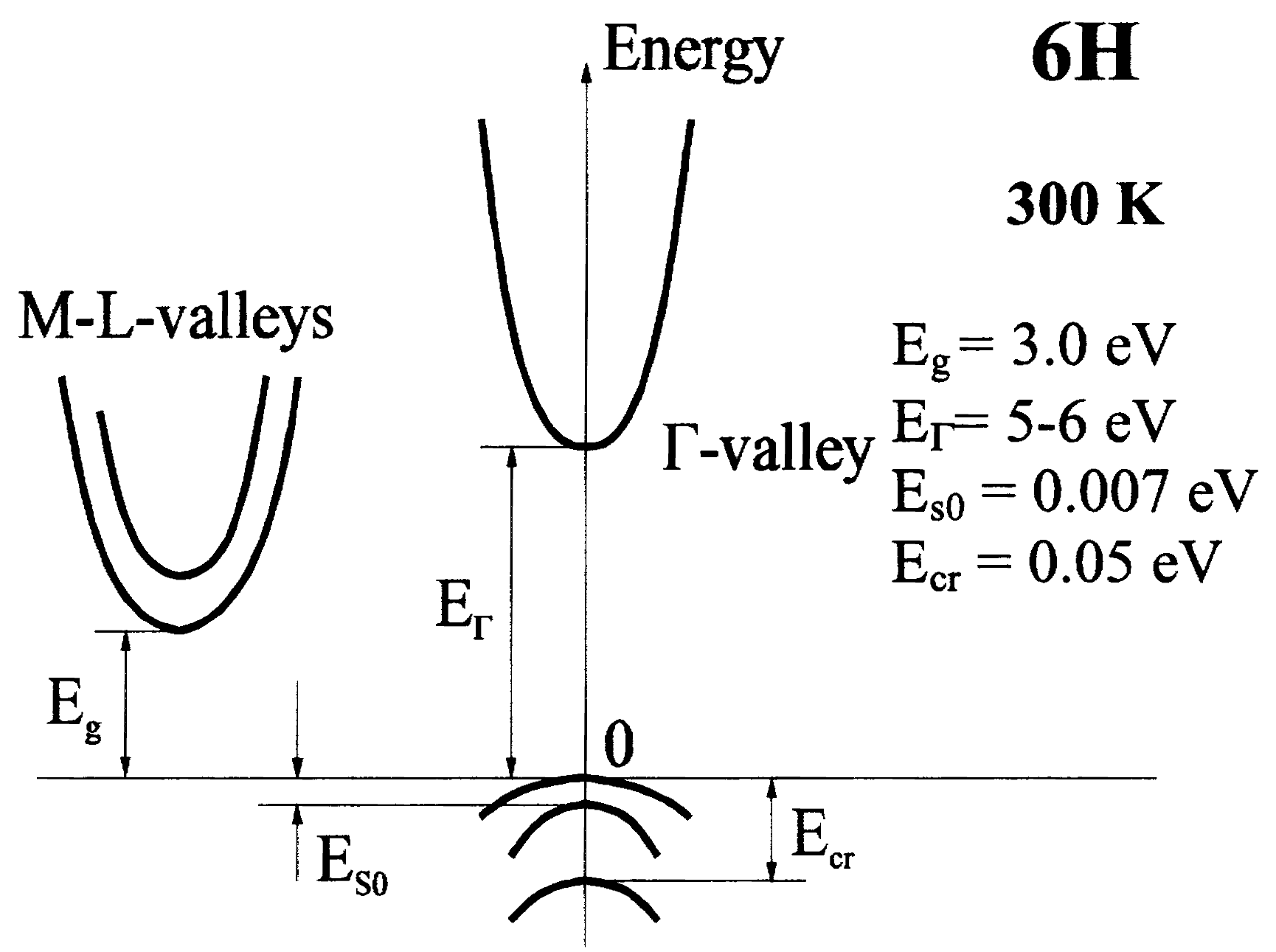
| 6H SiC | Moissanite-6H | ABCACB | ioffe.ru | Hexagonal | PL97 | Γ - M-L | 3.00 (300K) |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hyperfine parameters
Vasp manual on hyperfine calculations. Derive NGYROMAG parameters from Magnetogyric ratio (MR) of webelements.com.
NGYROMAG = MR[Mhz T-1 rad] / 2*PI
| Isotope | MR | NGYROMAG |
|---|---|---|
| 13 C | 67.283 | 10.7084 |
| 14 N | 19.338 | 3.0777 |
| 15 N | -27.126 | -4.3172 |
| 17 O | -36.264 | -5.772 |
| 19 F | 251.662 | 40.052 |
| 29 Si | -53.190 | -8.4655 |
| 31 P | 108.394 | 17.2514 |
| 69 Ga | 64.389 | 10.2477 |
| 71 Ga | 81.812 | 13.0207 |
| 73 Ge | -9.3603 | -1.4897 |
Core levels calculation
VASP manual: http://cms.mpi.univie.ac.at/vasp/vasp/ICORELEVEL_tag_core_level_shifts.html
Two main methods
1. Initial state approximation
ICORELEVEL = 1
2. Final state approximation
ICORELEVEL = 2
CLNT = ns # ns: species number
CLN = N # N: main quantum numer of excited core electron
CLL = L # L: l quantum number of excited core electron
CLZ = Z # Z: excited electron count (1 or 0.5)*
According Slater-Janak transition state approach (PRB 63 (2001) 205419; PRB 18 (1978) 7165)
Example: calculate C1s core level
ICORELEVEL = 2 CLNT = 1 # The first species is Carbon CLN = 1 # 1s core level, N=1 CLL = 0 # 1s core level, l=0 CLZ = 0.5 # 1/2
Generating Supercells
You can generate supercells from primitive cells by the wsgen package:
git://github.com/hornos/wsgen.git cd wsgen/src make
Copy src/wsgen and scgen to $HOME/bin . Edit the config file for the input generator. The scale variable sets multipliers for the lattice vectors and shift sets a shift for each sub-lattice (eg.: center sub-lattices for cubic or hexagonal place).
VASP
./scgen -p vasp -d -i <INPUT> -m "<LIST>"
where <INPUT> it the primitive CONTCAR and "<LIST>" is a space separated list of species for merge order of sub-lattices (eg.: "Si C").
You can make transformation on the merged geometry by creating a transform file merge.tf. Eg.: center the final geometry on the 1st Si atom (VMD atom index is used):
@center 1 Si
The final geometry is in merge.transform.POSCAR . Please be aware that the generated geometry is not VASP-normalized and not all atom is in the primitive cell of the supercell.
Brillouin Zone
K-points
| Lattice | X | L | M | W | K | A | H | N | P |
|---|---|---|---|---|---|---|---|---|---|
| FCC (cubic) | 1/2, 1/2, 0 | 1/2, 1/2, 1/2 | 1/4, 1/2, 3/4 | 3/8, 3/8, 3/4 | |||||
| Hexagonal | 0, 1/2, 1/2 | 0, 1/2, 0 | 1/3, 1/3, 0 | 0, 0, 1/2 | |||||
| BCC | -1/2, 1/2, 1/2 | 0, 0, 1/2 | 1/4, 1/4, 1/4 |